drawing 3d representations of molecules
Thoughts and Theory
Basic Molecular Representation for Machine Learning
From SMILES to Word Embedding and Graph Embedding
![]()

Machine learning has been applied to many issues in cheminformatics and life science, for example, investigating molecular property and developing new drugs. One critical issue in the problem-solving pipeline for these applications is to select a proper molecular representation that featurizes the target dataset and serves the downstream model. Figure 1 shows a conceptual framework for different molecular representations. Usually, a molecule is represented by a linear form as a SMILES string, or by a graph form as an adjacent matrix maybe together with a node aspect matrix for atoms and an edge attribute matrix for bonds. A SMILES cord could be further converted into different formats such as molecular fingerprint, i-hot encoding, or word embedding. On the other manus, the graph form of molecular representation could be directly used past the downstream model or exist converted into graph embedding for the task.

This mail describes some codes used in the implementation of the above conceptual framework, including:
- reading, drawing, analyzing a molecule,
- generating molecular fingerprint from a SMILES string,
- generating one-hot encoding from a SMILES string,
- generating word embedding from a SMILES string, and
- generating molecular representation in graph.
Reading, Drawing, and Analyzing a Molecule
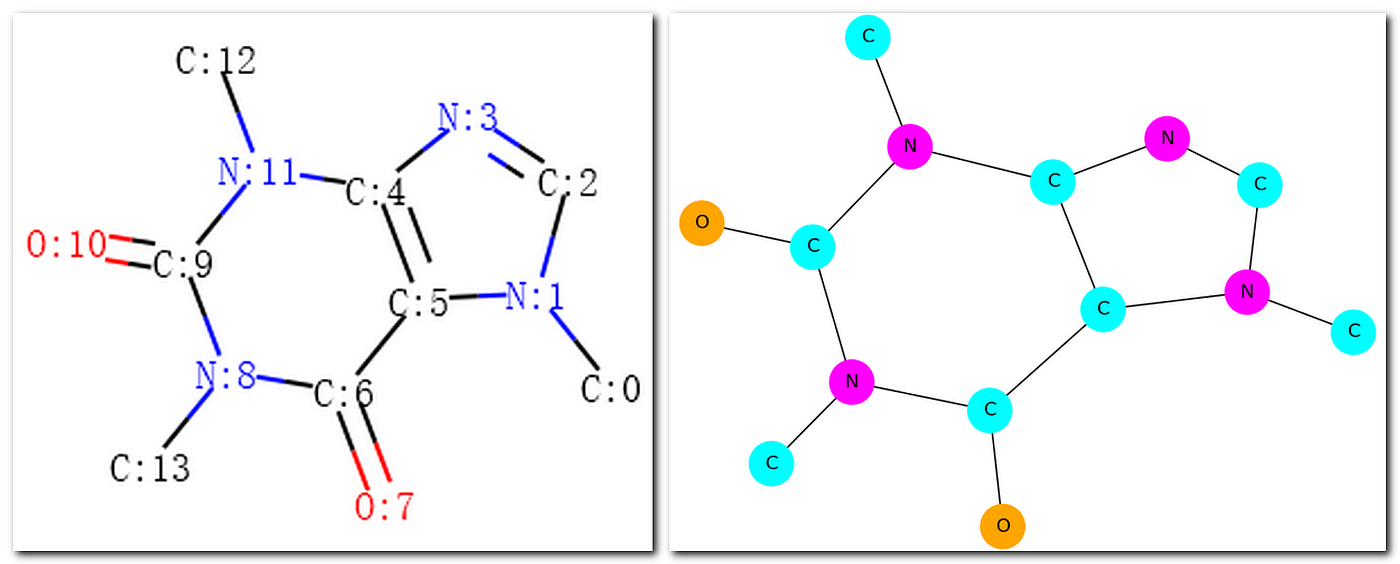
RDKit is an open up-source library for cheminformatics. Figure 2 shows the lawmaking for reading the SMILES string of caffeine and cartoon its molecular structure. Notice that C is carbon, N is nitrogen, and O is oxygen in a SMILES cord. A molecule could be displayed without labeling carbon as shown in Figure three or with labeling carbon as shown in Figure 4.


Figure 5 shows the code for displaying the atoms and bonds in the molecule of caffeine.
Figure 6 and Effigy 7 show the details of atoms and bonds in the molecule of caffeine, respectively. Notes:
- The term "aromatic" could be merely regarded as "ring" in the following tables. GetIsAromatic in Effigy half-dozen indicates if the atom is in a band or not, and GetBondType in Effigy vii indicates if the bail is in a band or not.
- Effigy 6 and Figure seven could be regarded every bit a simple atom attribute matrix and a simple bail attribute matrix in the conceptual framework shown in Figure i.
- The list of bonds in Figure vii could represent the graph class for a molecule, i.e., the link listing of an adjacency matrix.
Generating Molecular Fingerprint from a SMILES String
RDKit supports several fingerprint functions, which outputs could exist used for calculating molecular similarity or as the inputs to the downstream machine learning models. Figure 8 shows the codes for retrieving RDKit Fingerprint and Morgan Fingerprint, and Figure 9 shows the results of these fingerprint functions.
Generating One-Hot Encoding from a SMILES string
Considering SMILES strings as text in natural language, probably the simplest representation method for SMILES strings is one-hot encoding at the graphic symbol level. Figure 10 shows the lawmaking for generating one-hot encoding at the character level of a SMILES string.
Note that one-hot encoding could be also used at the atom level or in the atom/bail attribute matrix.
Generating Discussion Embedding from a SMILES String
In the context of language modeling, a more sophisticated approach for generating molecular representation is to apply the method of give-and-take embedding to the substructures of a molecule. The code in Figure eleven shows the procedure of using mol2vec and word2vec on generating word embedding for all the molecules in the HIV dataset. There are 41127 molecules in the dataset (Effigy 12) and each molecule is encoded as a 300-dimensional vector (Figure 13). Note that the lawmaking is extracted from "Simple ML In Chemistry Inquiry: RDkit & mol2vec" which explains the solution for predicting HIV activity in detail.
Generating Molecular Representation in Graph
The procedure of manipulating molecules/atoms/bonds in RDKit provides the foundation for generating the graph form of molecular representation. Effigy v, Effigy 6, and Effigy vii above accept shown the adjacency matrix, the node aspect matrix, and the edge aspect network for caffeine. Notwithstanding, converting a molecule in RDKit into a graph in NetworkX (an open-source library for network analysis) could leverage the research of the traditional graph algorithms and the mod graph models for investigating molecular structure and property. Figure xiv shows the lawmaking for converting a molecule in RDKit into a graph in NetworkX. Figure 15 shows the molecular graphs drawn past RDKit and NetworkX.
One important research area in graph networks is graph embedding. More often than not speaking, graph embedding consists of three topics: node-level embedding (which encodes nodes in a graph equally vectors), edge-level embedding (which encodes edges in a graph as vectors), and graph-level embedding (which encodes a whole graph as a vector.) In this post, we consider the term graph embedding as graph-level embedding, which finds a vector for a molecule that could be used as the input for the downstream models. Figure 16 shows the code for converting molecules in RDKit to graphs in NetworkX, and generating its graph embeddings via Graph2Vec under KarateClub. Graph2Vec is a graph embedding algorithm and KarateClub is a parcel providing unsupervised machine learning models for graph data. Figure 17 shows Graph2Vec embedding for the molecules in the HIV dataset. KarateClub has covered several graph embedding algorithms in the library.
Conclusions
This post has described several molecular representations, including cord-based format, graph-based format, and some variants such as word embedding and graph embedding. These molecular representations, together with dissimilar machine learning algorithms including deep learning models and graph neural networks, could serve as the baseline for approaching molecular machine learning problems.
Thanks for reading. If you take any comments, please feel costless to drop me a notation.
pattonintentookey.blogspot.com
Source: https://towardsdatascience.com/basic-molecular-representation-for-machine-learning-b6be52e9ff76
0 Response to "drawing 3d representations of molecules"
Post a Comment